최근 FDA는 바이오의약품 개발 시 비인간 영장류(NHP)의 3개월 초과 독성시험을 면제할 수 있는 혁신적인 가이드라인을 발표했습니다. 이는 종합증거(WoE)와 신기술 기반 대체법을 활용하여 신약 개발의 속도를 높이고 비용을 절감하는 핵심 전략입니다. 본 포스팅에서는 연구 현장에서 즉시 적용 가능한 규제 배경과 실무 가이드를 제공합니다.
성공적인 글로벌 IND 승인을 위해 NHP 독성시험 면제 조건과 최신 규제 동향을 반드시 숙지하세요.
인사이트 키워드: NHP독성시험면제, 3개월초과시험, 종합증거(WoE), 대체시험법(NAMs)
2025년 FDA 비임상 규제 변화와 NHP 독성시험 면제
신약 개발을 리드하는 벤처 연구원 및 팀장이라면 최근 비임상 규제 동향을 반드시 파악해야 합니다. 2025년 FDA가 발표한 단클론항체(Monoclonal Antibodies) 관련 지침은 업계에 큰 변화를 가져왔습니다. 핵심은 장기 독성 시험의 의무를 합리적으로 완화하는 것입니다.
규제 변화의 배경: 3R 원칙과 대체시험법(NAMs)
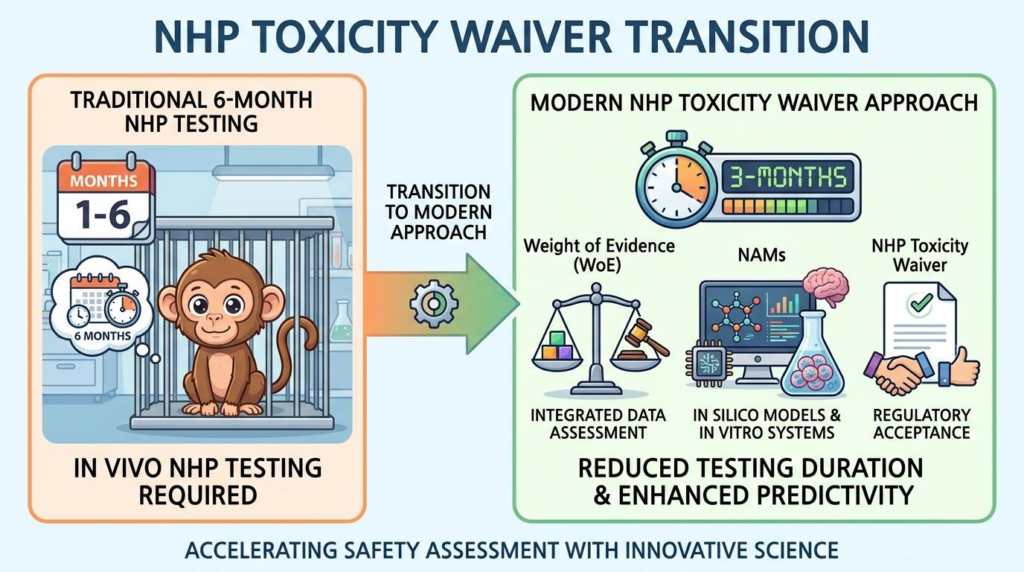
과거에는 만성 질환 치료제 허가를 위해 6개월 이상의 비인간 영장류(Non-Human Primate, NHP) 반복투여 독성시험(Repeated-dose toxicity study)이 필수적이었습니다. 하지만 최근 실험동물 보호를 위한 3R 원칙(Replacement, Reduction, Refinement)이 강력하게 대두되었습니다.
이에 따라 대체시험법(NAMs)의 신뢰성이 크게 향상되었습니다. 규제 기관은 기계적인 동물 실험 연장 대신, 과학적 데이터를 종합하여 안전성을 입증하는 유연한 방식을 권장하고 있습니다.
FDA 2025 가이드라인과 3개월 초과 시험의 규제적 의미
비임상 규제 문서에서 3개월 초과 시험이라는 단어는 단순한 수사가 아닙니다. 이는 명확한 법적, 제도적 기준을 의미합니다.
왜 3개월 초과 시험이 기준점이 되나요?
규제 문서에서 3개월 초과 시험이라는 표현을 명시적으로 사용하는 이유가 있습니다. 이는 국제의약품규제조화위원회(ICH)의 S6(R1) 및 M3(R2) 가이드라인에서 비임상 시험 기간을 나누는 법적 기준이 바로 ‘3개월(13주)’이기 때문입니다.
- 3개월 이하: 임상 1상과 2상을 지원하기 위해 반드시 수행해야 하는 단기 및 아만성(Subchronic) 시험입니다.
- 3개월 초과에서 6개월: 품목 허가를 지원하는 장기 및 만성(Chronic) 시험 영역입니다.
즉, 3개월까지의 초기 시험에서 중대한 독성 신호(Toxicological Signals)가 관찰되지 않는다면, 그 이후 추가적인 동물 실험을 면제받을 수 있습니다. 모호함을 없애기 위해 ‘3개월 초과’라는 정량적 단어를 사용하여 규제적 분수령을 명확히 한 것입니다.
| 구분 | 투여 기간 (Timeline) | 요구 사항 | 생략 가능성 |
|---|---|---|---|
| 3개월 이하 시험 | 0개월에서 3개월 | 반드시 실제 동물 시험 수행 필수 | 낮음 |
| 3개월 초과 시험 | 3개월 이후에서 6개월 | WoE(종합증거) 충족 시 장기 시험 대체 | 높음 |
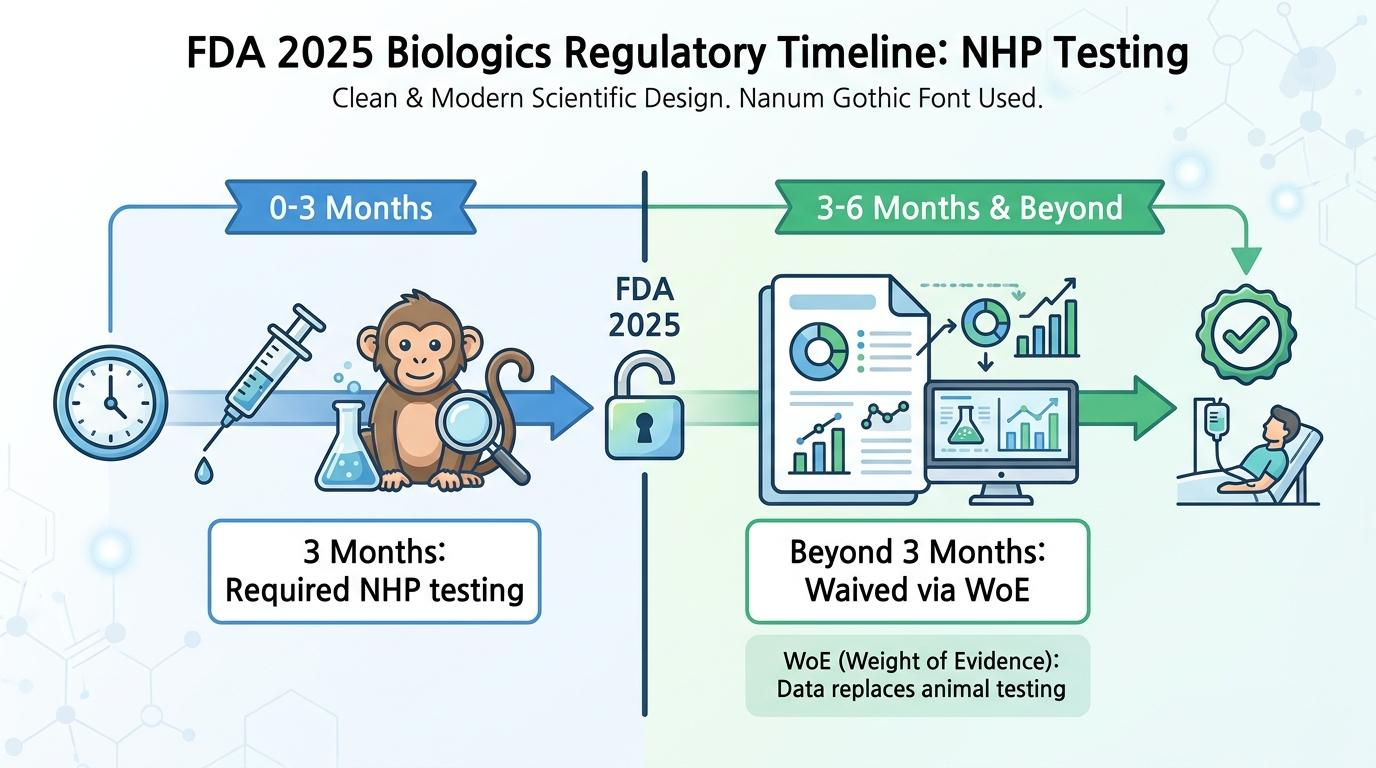
[그림 1] 규제 타임라인에 따른 비임상 시험 요구 사항 변화
3개월을 기준으로 나뉘는 비임상 규제 변화입니다. 0에서 3개월 구간은 필수적인 동물 시험 구간이지만, 3개월 초과 구간부터는 종합증거(WoE)를 적극 활용하여 추가적인 영장류 장기 시험을 효과적으로 대체할 수 있음을 나타냅니다.
2025년 FDA 단클론항체 지침과 3R 원칙의 적용
2025년 FDA가 발표한 단클론항체 초안 지침은 비임상 트렌드의 전환점입니다. 비설치류, 특히 NHP에서 3개월을 초과하는 시험은 일반적으로 불필요하다고 명시했습니다.
대신 3개월 시험 결과와 종합증거(Weight of Evidence, WoE)를 결합하세요. 위험 평가를 통해 장기 안전성을 충분히 입증한다면, 6개월 시험을 과감하게 생략하거나 축소할 수 있습니다. 이는 실험동물 보호를 위한 3R 원칙과 신기술 기반 대체시험법(NAMs) 활용을 강력하게 뒷받침합니다.
실무 팁(Pro-tip): 신약 개발 단계에서 3개월 독성시험을 기획할 때, 단순히 생존율이나 기본 병리 데이터만 수집하지 마세요. 추후 NHP 독성시험 면제를 위한 WoE 자료로 활용할 수 있도록, 초기 단계부터 면역원성 데이터와 기전적 바이오마커를 함께 분석하는 것이 유리합니다.
3개월 초과 시험 면제를 위한 종합증거(WoE) 구성 요소
성공적인 NHP 독성시험 면제를 위해서는 단순한 시험 생략이 아닌, 과학적으로 타당한 근거 제시가 필수적입니다. 규제 기관을 설득하기 위해 개발사가 제출해야 하는 종합증거(WoE)의 핵심 요소는 다음과 같습니다.
- 초기 독성 데이터 (1에서 3개월): 가장 기본이 되는 자료입니다. 초기 시험 구간에서 중대한 독성 신호(Toxicological Signals)나 예상치 못한 면역원성 이슈가 없음을 증명해야 합니다.
- 신기술 기반 대체시험법 (NAMs): 생체 외(In vitro) 데이터를 적극 활용하세요. 컴퓨터 시뮬레이션(전산독성학)이나 오가노이드 모델 등을 통해 표적 독성 및 오프타겟(Off-target) 효과를 검증합니다.
- 기존 학술 및 임상 데이터: 타겟이 동일하거나 작용 기전이 유사한 기승인 약물의 비임상 및 임상 데이터를 참고 자료로 활용하여 안전성을 뒷받침합니다.
- 표적 특성 및 작용 기전: 장기 투여 시 인체 내에서 만성 부작용을 유발할 가능성이 구조적, 기전적으로 낮다는 점을 논리적으로 소명해야 합니다.
NHP 독성시험 면제의 기대 효과: 시간과 비용의 혁신
이러한 규제 혁신은 연구 현장에 실질적인 이점을 제공합니다. 가장 먼저, 비임상 시험에 소요되는 기간이 수개월 단축되어 임상(IND) 진입 속도가 비약적으로 빨라집니다. 또한, 가장 고가의 비용이 발생하는 영장류 장기 시험을 생략함으로써 연구 개발 예산을 크게 절감할 수 있습니다.
WoE 구성을 위한 필수 결합 분석 데이터 확보하기
설득력 있는 WoE를 구성하려면 약물과 표적 간의 결합 특성을 정밀하게 분석하는 것이 중요합니다. 신뢰할 수 있는 실시간 상호작용 데이터를 확보하여 규제 기관을 효과적으로 설득하고 싶다면 다음 분석법을 참고하세요.
세포 수준에서의 결합 친화도를 입증하여 NAMs 데이터를 보완해야 하나요? 생체 내 환경과 유사한 분석 결과가 필요하다면 아래 링크를 확인하세요.
비임상 시험 전략 수립이나 맞춤형 분석 서비스 도입에 대해 전문가의 조언이 필요하신가요? 단편적인 실험을 넘어 IND 승인을 위한 통합 데이터를 구축하세요. 지금 바로 상담을 신청하여 신약 개발의 성공률을 높이세요.
맞춤형 비임상 분석 문의하기자주 묻는 질문 (FAQ)
Q. 3개월 초과 시험 면제는 모든 바이오의약품에 무조건 적용되나요?
아닙니다. 현재는 주로 단클론항체와 같이 타겟 특이성이 명확한 모달리티에 우선적으로 적용됩니다. 개발 중인 약물의 기전과 위험성에 따라 FDA와의 사전 협의(Pre-IND meeting)가 필수적입니다.
Q. 종합증거(WoE) 보고서는 언제부터 준비해야 하나요?
후보물질 최적화 및 초기 비임상 기획 단계부터 선제적으로 준비하세요. 사후에 데이터를 끼워 맞추는 것이 아니라, 처음부터 NAMs와 결합 데이터를 염두에 두고 디자인해야 합니다.
Q. 신기술 대체시험법(NAMs) 데이터만으로 동물시험을 완전히 대체할 수 있나요?
아직 완전한 대체는 어렵습니다. 현재 규제 가이드라인은 초기 1에서 3개월의 필수 동물시험 데이터를 기반으로, 이후의 장기 시험을 NAMs로 ‘보완 및 대체’하는 것을 권장합니다.
핵심 용어 정리 (Glossary)
- WoE (Weight of Evidence): 종합증거. 특정 가설을 입증하기 위해 이용 가능한 모든 과학적 데이터를 종합적으로 평가하는 접근법입니다.
- NAMs (New Approach Methodologies): 신기술 기반 대체시험법. 컴퓨터 모델링, 장기 칩(Organ-on-a-chip), 오가노이드 등 동물실험을 대체하거나 줄일 수 있는 혁신 기술을 뜻합니다.
- 3R 원칙: 실험동물의 사용을 대체(Replacement), 감소(Reduction), 고통 완화(Refinement)하려는 국제적인 연구 윤리 가이드라인입니다.
연관 토론 주제
- 이중항체(Bispecifics) 개발 시 NHP 면역원성 데이터의 해석 한계와 대안
- 오가노이드 기반 NAMs 데이터가 FDA 허가 자료로 인정받기 위한 밸리데이션 요건
- 전산독성학(In silico) 모델을 활용한 비임상 후보물질 초기 스크리닝 전략
주요 참고문헌
- FDA Draft Guidance (2025). “Monoclonal Antibodies: Streamlined Nonclinical Safety Studies”.
- ICH S6(R1) Guideline. “Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals”.
- ICH M3(R2) Guideline. “Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals”.
문의 QR 코드 (메시지 연결)
* 본 포스팅에 언급된 기관명(FDA, ICH 등) 및 관련 제도는 해당 기관의 등록 상표 및 공식 가이드라인을 참조하였으며, 본 콘텐츠는 정보 제공을 목적으로 작성되었습니다.